Article Type: Case Report, Volume 3 Issue 1
*Corresponding author: Amrit Kaur Kaler
Department of Molecular Pathology and Genomics, Kokilaben Dhirubhai Ambani Hospital and Medical Research Institute, Mumbai, Maharashtra, India.
Email: amrit_kaler@yahoo.co.in
Received: Dec 06, 2025 Accepted: Dec 29, 2025 Published: Jan 06, 2026
Citation: Kaler AK, Shaikh IM, Manoharan R, Jariwala K, Kulkarni Y, et al. Germline CHEK2 and lynch like phenotype in a rare metachronous presentation as haematological and gynaecological malignancies. Ann Case Rep Med Images. 2026; 3(1): 1058.
Copyright: Kaler AK et al. © All rights are reserved
A germline CHEK2 nonsense variant was identified in a patient presenting with both diffuse large B-cell lymphoma, a subtype of non-Hodgkin’s lymphoma, and endometrioid adenocarcinoma, through next-generation sequencing of a 113-gene hereditary cancer panel. This truncating mutation, typically associated with breast and colorectal cancers, suggests an expanded cancer predisposition spectrum. The endometrial tumor demonstrated loss of MLH1 and PMS2 expression, resembling a Lynch-like phenotype. This unique combination of haematologic and solid tumors in the presence of a pathogenic CHEK2 variant highlights the gene’s broader role in hereditary cancer syndromes. The findings support the utility of comprehensive germline testing and genetic counseling in patients with complex cancer histories.
Keywords: CHEK2 positive; Endometrioid adenocarcinoma; DLBCL; Lynch-like; Pembrolizumab.
Diffuse Large B-Cell Lymphoma (DLBCL) is an aggressive subtype of B-cell non-Hodgkin lymphoma, which comprises a diverse group of disorders characterized by distinct pathological, clinical, and molecular features. According to the WHO, the DLBCL is the most prevalent subtype of lymphoid neoplasm, accounting for around 40% of all Non-Hodgkin’s Lymphomas (NHLs) worldwide [5]. Histologically characterized by diffuse proliferation of large neoplastic B lymphoid cells with a nuclear size equal to or exceeding normal histiocyte nuclei [7]. Established risk factors include immune system compromise, such as HIV/AIDS, organ transplantation, or inherited immunodeficiencies as well as viral infections (e.g., Epstein-Barr virus, hepatitis C and B), autoimmune disorders, obesity, environmental exposures, and underlying genetic predisposition [11].
Endometrioid carcinoma is the most common histological subtype of endometrial cancer, typically arising in the setting of prolonged unopposed estrogen exposure and often evolving from endometrial hyperplasia with atypia. Histologically, it resembles proliferative-phase endometrial glands and generally carries a favorable prognosis when diagnosed at an early stage [21]. Following germline testing, a pathogenic CHEK2 mutation was detected, supporting the growing evidence that CHEK2 predisposition may extend beyond traditionally recognized cancers.
The checkpoint kinase 2 (CHEK2) gene (OMIM 604373) coding for CHEK2 protein has been reported as a moderate-penetrance, multi-organ cancer susceptibility gene whose alterations increase the risk of different malignancies including breast, colorectal or prostate cancers [12]. CHEK2 is a nuclear serine/ threonine kinase contributing to the ATM CHEK2 p53 cascade, a part of the DNA damage response (DDR) system. The CHEK2 is activated by ATM-mediated phosphorylation [14] and in turn, phosphorylates several effector proteins involved in regulation of the cell cycle and apoptosis upon DDR.
CHEK2-associated cancer predisposition is inherited in an autosomal dominant pattern. Most affected individuals are heterozygous for a pathogenic CHEK2 variant, although biallelic pathogenic variants are observed infrequently. In the majority of cases, the heterozygous variant is inherited from a parent, regardless of the parent’s cancer history. Offspring of individuals with a heterozygous CHEK2 pathogenic variant have a 50% risk of inheriting the variant. In cases where an individual carries biallelic pathogenic variants, offspring is expected to inherit at least one pathogenic variant. Once a pathogenic CHEK2 variant is identified in a proband, predictive genetic testing can be offered to at-risk relatives to guide cancer surveillance and risk management strategies [23].
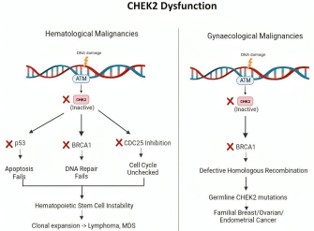
Figure 1: CHEK2 pathway - DNA damage activates the ATM–CHK2 signalling pathway, leading to phosphorylation of p53, BRCA1, CDC25A/C, and E2F1. These downstream effectors coordinate responses including cell cycle arrest, homologous recombination repair, and apoptosis.
The metachronous occurrence of Diffuse Large B-Cell Lymphoma (DLBCL) followed by endometrioid carcinoma is exceptionally rare, with only a few documented cases reported in the literature. We present the case of a 65-year-old female who was initially diagnosed with DLBCL, a high-grade subtype of nonHodgkin’s lymphoma, and subsequently developed endometrioid adenocarcinoma during clinical follow-up. The patient’s family history is notable for malignancies in multiple first-degree relatives, including breast and renal cancers. This sequential development of distinct primary tumors raises strong suspicion for an underlying hereditary cancer predisposition syndrome. The case highlights the importance of maintaining a high index of suspicion for genetic susceptibility in patients presenting with multiple primary malignancies, particularly when accompanied by a significant familial cancer history.
An initial genetic counselling session was conducted as part of the patient’s clinical evaluation to assess the likelihood of a hereditary cancer syndrome. Genetic counselling is a structured communication process that provides individuals and families with information about the medical, psychological, and familial implications of genetic contributions to disease. It involves interpreting family and medical histories to assess the chance of disease occurrence or recurrence, educating about inheritance patterns, testing options, and management, and facilitating informed decision-making [9,13]. In this case, counselling focused on evaluating the patient’s personal cancer history, reviewing the family pedigree, and identifying potential patterns suggestive of inherited cancer risk. Given the findings from this evaluation, germline genetic testing was recommended to investigate the presence of inherited variants associated with cancer susceptibility.
Germline genetic testing is a molecular diagnostic approach that analyses germline DNA to detect inherited genetic alterations that may increase disease risk. For this patient, testing was performed using a targeted Next-Generation Sequencing (NGS) panel that included 113 genes implicated in hereditary cancer predisposition. These genes were selected based on established associations with a range of cancer types and syndromes. Peripheral blood was collected, and genomic DNA was extracted using standardized protocols. Library preparation was conducted using the Nextera DNA Flex library preparation kit, and sequencing was performed on the Illumina MiSeq platform, which enables high-throughput, short-read sequencing.
The assay was designed to provide comprehensive variant detection, including Single Nucleotide Variants (SNVs), small insertions and deletions (indels), Copy Number Variations (CNVs), and structural rearrangements. Raw sequencing data underwent quality control, alignment, and variant calling using validated bioinformatics pipelines. All identified variants were interpreted in accordance with the guidelines established by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP), which provide a standardized framework for classifying variants based on pathogenicity [20].
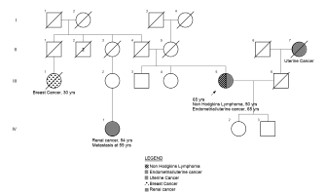
Figure 2: Pedigree of a multi-generational family with a history of diverse malignancies - The diagram illustrates affected individuals across three generations with various cancers, including breast cancer (diagnosed at age 30), uterine cancer, kidney cancer (diagnosed at age 54; metastasis at 59), and Non-Hodgkin’s lymphoma (diagnosed at age 65). One female individual was diagnosed with both uterine cancer (age 50) and Non-Hodgkin’s lymphoma (age 65). Affected individuals are indicated by filled symbols, with distinct cross-hatching patterns representing different cancer types. Deceased individuals are marked with a diagonal slash. The proband is indicated by an arrow. Clinical details such as age at diagnosis (dx.) and metastasis (mets.) are annotated where available. The familial clustering and early age of onset suggest a potential underlying hereditary cancer predisposition syndrome.
A 65-year-old female patient was initially diagnosed with diffuse large B-cell lymphoma (DLBCL), a subtype of non-Hodgkin’s lymphoma. At the time of diagnosis, a specimen from the portal lymph node was submitted for histopathological examination. The specimen consisted of one lymph node measuring 1.5×1.5 cm, which was entirely processed. Additionally, fragmentary pieces of soft tissue collectively measuring 3×3 cm were received and also entirely processed for microscopic evaluation. Following this diagnosis, the patient completed six cycles of R-CHOP (Rituximab-Cyclophosphoamide, Doxoribucin, Vincristine, Prednisone) chemotherapy, followed by consolidative radiotherapy, achieving initial remission. However, several years later, during routine clinical follow-up, she presented with newonset symptoms. Subsequent investigations led to a diagnosis of endometrioid adenocarcinoma, thereby establishing the presence of a metachronous malignancy.
The patient’s family history is notable for a multi-generational pattern of diverse malignancies, raising clinical suspicion for a hereditary cancer predisposition syndrome. Affected relatives across three generations were reported to have developed a range of cancers, including breast cancer diagnosed at age 30, uterine cancer and kidney cancer diagnosed at age 54 with metastasis at age 59. The proband (index patient) also fits this pattern, having developed DLBCL and later endometrioid adenocarcinoma.
As part of her ongoing evaluation, the patient underwent comprehensive germline genetic testing using a 113-gene hereditary cancer panel via targeted next-generation sequencing (NGS). This revealed a heterozygous nonsense variant in the CHEK2 gene (NM_007194.4): chr22:29130652G>A, c.58C>T, p.Gln20Ter, located in exon 2. This substitution introduces a premature stop codon at amino acid position 20, producing a truncated protein of only 19 amino acids compared to the fulllength 543-residue kinase. The truncated protein is predicted to be non-functional due to the loss of all major functional domains, and the mutant transcript is likely subjected to nonsense-mediated mRNA decay (NMD), resulting in markedly reduced or absent protein expression.
The CHEK2 variant was detected in 48.58% of total sequencing reads (1021 reads), consistent with a heterozygous allelic state. This variant has been catalogued in the dbSNP database (rs536907995) and is observed at a low frequency (0.006%) in population datasets such as gnomAD, with 102 reported heterozygotes, indicating that it is a rare variant in the general population. The variant is classified as pathogenic/likely pathogenic in ClinVar (VCV000133887.40) based on multiple submissions linking it to hereditary breast and ovarian cancer syndromes.
The CHEK2 gene encodes a serine/threonine kinase that plays a central role in the DNA damage response by phosphorylating several key proteins such as BRCA1, TP53, CDC25A, and CDC25C, thereby facilitating cell cycle arrest and DNA repair in response to genotoxic stress. Loss-of-function variants in CHEK2 impair genomic stability and are associated with increased cancer susceptibility. This truncating CHEK2 variant is known to follow an autosomal dominant pattern of inheritance, with a 50% probability of transmission to first-degree relatives. Female carriers are at significantly increased risk for breast cancer, and there is growing evidence linking CHEK2 variants to ovarian and other cancers.
The variant’s pathogenicity is supported by its previous identification in multiple ethnically diverse cohorts affected by breast and ovarian cancers, as well as by functional studies showing that truncated CHEK2 proteins fail to maintain checkpoint fidelity in response to DNA damage. The test showed high-quality sequencing metrics, with an average depth of coverage of 888x, and 97.53% of target regions covered at ≥30x, ensuring high confidence in variant detection. Over 99.61% of reads aligned to the hg19 reference genome, and 94.7% of bases achieved a quality score ≥Q30, reflecting high sequencing accuracy. The technical parameters confirm the test’s ability to reliably detect Single Nucleotide Variants (SNVs), indels, CNVs, and structural variants with high sensitivity and specificity.
Subsequent staging investigations with whole-body PET-CT revealed increased FDG uptake in the uterus, bilateral obturator, common iliac, and presacral lymph nodes, retrocaval and retroperitoneal regions, a subcapsular lesion in segment VII of the liver, multiple lung nodules, left inguinal level IV lymph node, and bilateral subpectoral lymph nodes. Imaging also showed a heterogeneous endometrial mass with cervical stroma and right parametrial extension, along with distant metastases including peritoneal deposits, lymph nodes above the level of the renal vein, and suspicious pulmonary metastases. Based on MRI findings, the patient was staged as FIGO stage IV C.
The patient is receiving immunotherapy with pembrolizumab and is responding well, with clinical evaluation indicating good treatment tolerance and a favorable therapeutic response. This case illustrates the clinical utility of germline genetic testing in uncovering potential hereditary cancer syndromes and emphasizes the importance of integrating family history, molecular diagnostics, and advanced imaging in the evaluation of patients presenting with multiple primary malignancies.
CHEK2 in haematologic malignancies
The presence of CHEK2 mutations in haematologic malignancies such as Non-Hodgkin’s Lymphoma (NHL) remains underexplored, though emerging evidence suggests a contributory role. While NHL predominantly arises from clonal expansions of B, T, or NK cells and is typically linked to aberrations in genes regulating apoptosis and immune function [1], recent germline analyses have identified CHEK2 variants in up to 7.4% of NHL patients [12]. These mutations, particularly truncating and missense protein-altering variants, were associated with a moderate increase in lymphoma risk (odds ratio ~2.86) and worse progression-free survival. Such findings suggest that CHEK2 may contribute to lymphomagenesis through genomic instability, aligning with its recognized tumor suppressor role [12,4].
CHEK2-driven tumorigenesis in MMR-deficient cancers
CHEK2, a well-established moderate-penetrance cancer susceptibility gene, was initially implicated in estrogen receptor positive breast cancer through recurrent founder mutations such as 1100delC and p.Ile157Thr. However, growing evidence from epidemiological and molecular studies has expanded its relevance to a broader range of solid tumors, including prostate [12], pancreatic cancers [19], etc. Consistent with its function as a tumor suppressor, CHEK2-associated malignancies often demonstrate Loss of Heterozygosity (LOH) at the wild-type allele, supporting a classical two-hit hypothesis. However, these tumors typically lack Homologous Recombination Deficiency (HRD), distinguishing them from BRCA1/2-driven cancers and potentially limiting the therapeutic efficacy of PARP inhibitors [22,23].
In this case, the patient presents with endometrioid carcinoma showing loss of MLH1 and PMS2 protein expression on Immunohistochemistry (IHC), a finding that indicates Mismatch Repair (MMR) deficiency at the tumor level. However, germline testing revealed no mutations in MMR genes, including MLH1, which effectively rules out classical Lynch syndrome. The patient also has a germline CHEK2 mutation, which, though not directly involved in MMR, contributes to genomic instability. The probable methylation of MLH1 gene contributed towards the progression of endometrioid adenocarcinoma. Furthermore, the patient’s prior history of Diffuse Large B-Cell Lymphoma (DLBCL), alongside the current diagnosis of endometrioid carcinoma, suggests an underlying hereditary cancer predisposition that extends beyond the typical Lynch syndrome spectrum. This constellation of findings MMR-deficient tumor, absence of germline MMR mutations, personal history of multiple malignancies, and a pathogenic CHEK2 variant is consistent with a Lynch-like syndrome, a clinical entity characterized by features of Lynch syndrome without detectable germline mutations in MMR genes. While a purely sporadic etiology in the context of a germline CHEK2 mutation and multiple primary cancers can be ruled out.
Hereditary cancer syndromes and CHEK2 in the HBOC spectrum
The patient’s clinical and familial cancer history suggests a potential underlying hereditary cancer syndrome that extends beyond the classical BRCA1/2-associated Hereditary Breast and Ovarian Cancer (HBOC) framework. While BRCA1/2 mutations account for a substantial proportion of HBOC cases, recent advances in multigene panel testing have identified moderate penetrance genes, such as CHEK2, that contribute to inherited cancer susceptibility and expand the phenotypic spectrum of HBOC-related malignancies [21,17]. Pathogenic variants in CHEK2 have been linked to an increased risk of breast, endometrial, and renal cancers [10,12], while non-Hodgkin’s lymphoma is not traditionally linked to CHEK2, multiple studies now report CHEK2 variants in 1.5%-7.4% of NHL patients including DLBCL, suggesting a broader oncogenic potential for this gene.
In endometrial cancer, the prevalence of CHEK2 mutations is low (<1%) in large unselected cohorts, but its presence has been detected in endometrioid carcinoma, a hormone-responsive tumor type [18]. While statistical associations with risk are weak or non-significant, the presence of CHEK2 mutations may represent an alternative oncogenic pathway involving impaired DNA repair. Some studies also suggest that CHEK2 may act as a polygenic modifier in combination with MMR gene dysfunction in Lynch-like presentations [16].
Clinical implications and future perspectives
The clinical and molecular features are indicative of a Lynchlike syndrome, with potential microsatellite instability or mismatch repair deficiency suggesting eligibility for immunotherapy. Pembrolizumab, a PD-1 immune checkpoint inhibitor, has demonstrated durable antitumor activity in MSI-H/dMMR tumors across various tissue types. The KEYNOTE-158 trial notably supported pembrolizumab’s tumor-agnostic approval based on its efficacy in non-colorectal MSI-H/dMMR cancers, including endometrial carcinoma [15]. In addition, the KEYNOTE-028 study highlighted clinical benefit in PD-L1 positive endometrial cancer [2]. Although diffuse large B-cell lymphoma is not typically associated with Lynch syndrome, its presence in this case may reflect broader genomic instability or a multifactorial predisposition. Given the patient’s clinical and familial features, immunotherapy with pembrolizumab should be considered, particularly if the tumor demonstrates MSI-H or dMMR status, aligning with current evidence for checkpoint inhibition in hereditary or Lynch-like malignancies [3].
This case broadens the phenotypic spectrum associated with CHEK2 mutations, illustrating a rare but clinically significant combination of haematologic and gynaecologic malignancies in a patient with a germline CHEK2 pathogenic variant. Although these cancer types are not traditionally linked to CHEK2, their co-occurrence in this context calls for a re-evaluation of the gene’s role in hereditary cancer predisposition, particularly in individuals with complex cancer histories and notable familial patterns of disease.
Significantly, this diagnosis followed a comprehensive genetic evaluation, beginning with genetic counseling to assess hereditary cancer risk based on personal and family history. The presentation bears resemblance to Lynch-like syndromes, where tumors characteristic of mismatch repair deficiency occurs without identifiable MMR gene mutations. This overlap suggests that defects in DNA repair genes such as CHEK2 may contribute to similar oncogenic pathways, warranting further study.
These findings highlight the importance of broad germline testing and detailed genetic assessment, especially in atypical clinical scenarios. Incorporating comprehensive gene panels and thorough pedigree analysis enhances diagnostic accuracy, informing personalized surveillance, and supporting cascade testing in families.
Patient consent: The authors certify that they have obtained all appropriate patient consent forms.